

بدخیمیهای روده کوچک، بدخیمیهای نادری هستند. تخمین زده میشود که در سال ۲۰۱۶، ۱۰،۰۹۰ نفر در ایالات متحده به این بیماری مبتلا شدهاند و ۱۳۳۰ نفر بر اثر این بیماری جان خود را از دست دادهاند. انواع بافتشناسی متعددی از بدخیمیهای اولیه روده کوچک وجود دارد که شایعترین آنها آدنوکارسینوما، کارسینوئید، سارکوم و لنفوم هستند. متاستاز از سایر انواع تومور مانند ملانوما، سرطان ریه، روده بزرگ و تخمدان نیز مشاهده میشود. این فصل بر شایعترین بدخیمیهای اولیه روده کوچک (آدنوکارسینوم، کارسینوئید، لنفوم و سارکوم) تمرکز خواهد کرد.
اپیدمیولوژی
میزان بروز سرطانهای روده کوچک در حال افزایش است. در ایالات متحده، میزان بروز سرطانهای روده کوچک از ۱۱.۸ مورد در هر میلیون نفر در سال ۱۹۷۳ به ۲۲.۷ مورد در هر میلیون نفر در سال ۲۰۰۴ افزایش یافته است (بیلیموریا و همکاران). آمریکاییهای آفریقاییتبار در ایالات متحده بالاترین میزان بروز سرطان روده کوچک را بر اساس سن در جهان دارند (هاسلکورن و همکاران). از نظر تاریخی، آدنوکارسینوما 30 تا 50 درصد از بدخیمیهای روده کوچک، کارسینوئید 20 تا 30 درصد و لنفومها و تومورهای استرومایی هر کدام 15 درصد را تشکیل میدهند. با این حال، مطالعهای توسط بیلیموریا و همکاران با استفاده از پایگاه داده ملی سرطان (NCDB، 1985-2005) و پایگاه داده نظارت اپیدمیولوژی و نتایج نهایی (SEER، 1973-2004) نشان داد که از 67843 بیمار شناسایی شده، 37.4 درصد کارسینوئید، 36.9 درصد آدنوکارسینوما، 17.3 درصد لنفوم و 8.4 درصد تومورهای استرومایی داشتند. افزایش میزان بروز تومورهای کارسینوئید در ایالات متحده از ۲.۱ در هر میلیون نفر در سال ۱۹۷۳ به ۹.۳ در هر میلیون نفر در سال ۲۰۰۴ بوده است که نشاندهنده افزایش ۳۴۰.۵ درصدی است. دلیل افزایش میزان بروز مشخص نیست، اما ممکن است به افزایش میزان تشخیص مربوط باشد.
هنگامی که به مناطق آناتومیک مختلف روده کوچک تقسیمبندی میشود، کارسینوئیدها در ایلئوم شایعتر هستند در حالی که آدنوکارسینومها بیشتر در روده کوچک پروگزیمال یافت میشوند. سارکومها و لنفومها معمولاً در روده کوچک دیستال دیده میشوند، اما میتوانند در سراسر آن یافت شوند.
عوامل خطر
روده کوچک ۷۵٪ از طول دستگاه گوارش (GI) را تشکیل میدهد، اما بدخیمیهای روده کوچک کمتر از ۵٪ از کل سرطانهای دستگاه گوارش را شامل میشوند.
عوامل محافظتی آزاد شده توسط روده کوچک، کیفیت مدفوع و سطح پایین عوامل سرطانزا ممکن است این اختلاف را توضیح دهند. یکی دیگر از عواملی که تصور میشود در بروز نسبتاً پایین سرطان روده کوچک نقش دارد، ترشح ایمونوگلوبولین A توسط روده کوچک است که نقش اساسی در ایمنی مخاطی دارد.
سندرمهای ارثی خانوادگی نیز با سرطان روده کوچک مرتبط هستند. این موارد شامل پولیپوز آدنوماتوز خانوادگی، پوتز-جگرز، سندرم لینچ، سندرم کودن، نوروفیبروماتوز فون رکلینگهاوزن و بیماری کرون است.
تظاهرات بالینی
بیماران مبتلا به بدخیمیهای روده کوچک در مقایسه با بیماران مبتلا به ضایعات خوشخیم، بیشتر احتمال دارد که با شکایات گوارشی مراجعه کنند. علائم شایع شامل درد شکم (75٪)، کاهش وزن (28٪)، انسداد روده (25٪) و خونریزی (24٪) است. سرطانهای دوازدهه در صورت درگیری آمپول میتوانند با زردی تظاهر کنند. تشخیص اغلب به تأخیر میافتد مگر اینکه تظاهر بیماری شامل یک وضعیت اورژانسی مانند انسداد، خونریزی یا سوراخ شدن باشد. سوراخ شدن روده علامت اولیه در 10٪ از بیماران است و بیشتر با لنفوم یا سارکوم دیده میشود.
بررسیهای تشخیصی
بررسیهای اولیه شامل گرفتن شرح حال جامع و معاینه فیزیکی است. در صورت وجود شک بالینی به تومور کارسینوئید، آزمایشهای آزمایشگاهی باید شامل شمارش کامل خون، الکترولیتهای سرم، آنزیمهای کبدی و 5-هیدروکسی ایندول استیک اسید (5-HIAA) باشد.
تصویربرداری مقطعی اغلب برای تشخیص استفاده میشود. تصویربرداری توموگرافی کامپیوتری (CT) حساسیت 80٪ برای ضایعات روده کوچک دارد و با سیتیاسکن یا امآرآی انتروگرافی به 85٪ تا 95٪ افزایش مییابد (پاپالاردو و همکاران و پیلول و همکاران). برای تومورهای کارسینوئید، سینتیگرافی اکتروئوتید آنالوگ سوماتوستاتین نشاندار شده با رادیو [111In-DTPA0] (OctreoScan) میتواند به عنوان یک روش کمکی برای شناسایی ضایعات اولیه و خارج شکمی استفاده شود که حساسیت 88٪ و ویژگی 97٪ دارد (پاپ و همکاران). آندوسکوپی امکان تشخیص و بیوپسی بدخیمیهای پروگزیمال روده کوچک در دوازدهه را فراهم میکند، با این حال ارزیابی ضایعات در روده کوچک دیستالتر چالش برانگیز است. انتروسکوپی تک بالون و دو بالون، روشهایی برای بررسی فراتر از رباط تریتز هستند.
مطالعهای توسط تاکانو و همکارانش، انتروسکوپی تک بالون و دو بالون را مقایسه کرد و میزان بالاتری از تکمیل انتروسکوپی کامل با دو بالون (0٪ در مقابل 57٪، p = 0.002) را نشان داد، اما هیچ تفاوتی در نتایج درمانی مشاهده نشد (تک بالون 28٪ در مقابل دو بالون 35٪، p = 0.63). استفاده از انتروسکوپی تک بالون یا دو بالون هنوز هم بر اساس ترجیح پزشک و در دسترس بودن آن است.
آندوسکوپی کپسول ویدیویی (VCE) روش دیگری است که به طور فزایندهای برای ارزیابی روده کوچک استفاده میشود. یک دوربین قرصی شکل بلعیده میشود و تصاویر کل روده کوچک را منتقل میکند. یک متاآنالیز ترکیبی توسط لوئیس و همکارانش، میزان کمتری از آسیبشناسی از دست رفته را در مقایسه با انتروسکوپی با میزان تکمیل معادل نشان داد.
VCE در بیمارانی که تخلیه معده آنها با تأخیر مواجه است، سابقه جراحی شکم دارند و آمادگی روده آنها ضعیف است، بازده تشخیصی ضعیفی دارد. خطرات VCE شامل احتباس کپسول است که در ۱٪ از بیماران نیاز به آندوسکوپی مجدد یا جراحی دارد.
با وجود ابزارهای تشخیصی متعدد موجود، تشخیص دقیق بدخیمی روده کوچک همچنان دشوار است و تنها در نیمی از موارد انجام میشود. هنگامی که هیچ تشخیصی انجام نشده است اما سوءظن بالینی بالا است و بیماران علائم خونریزی یا انسداد دستگاه گوارش دارند، باید لاپاراتومی اکتشافی انجام شود و آندوسکوپی حین عمل میتواند با کمک جراح انجام شود که امکان مشاهده کامل روده کوچک را فراهم میکند. لاپاروسکوپی تشخیصی همچنین میتواند در بررسی حفره صفاقی و گرفتن بیوپسی در صورت عدم وجود عوارض دستگاه گوارش کمک کند.
انواع خاص بدخیمیهای روده کوچک
آدنوکارسینوم
پاتولوژی
آدنوکارسینومهای روده کوچک (SBA) از نظر تاریخی 30٪ تا 50٪ از کل بدخیمیهای روده کوچک را تشکیل میدادند، اما مطالعات معاصر نرخ 36.9٪ را نشان دادهاند که به دومین بدخیمی اولیه شایع روده کوچک پس از تومورهای کارسینوئید کاهش یافته است. بیش از نیمی از این ضایعات در دوازدهه و پس از آن ژژونوم و ایلئوم ایجاد میشوند. درست مانند آدنوکارسینوم کولورکتال، این تومورها میتوانند بزرگ شوند و از طریق سروز به ساختارهای مجاور حمله کنند. بزرگ شدن تومور میتواند باعث علائم انسدادی یا در صورت ابتلا به دوازدهه اولیه، انسداد خروجی معده شود. نقض لایه مخاطی میتواند منجر به خونریزی دستگاه گوارش یا کمخونی مزمن شود.
تجزیه و تحلیلهای ژنتیکی، جهشهایی را در چندین ژن کلیدی نشان میدهد. ژن پولیپ آدنوماتوز کولی (APC) در 10 تا 18 درصد از SBA در مقایسه با 80 درصد از سرطانهای کولورکتال پراکنده (CRC) جهش یافته است (Wheeler و همکاران، Arai و همکاران). اگرچه APC در هر دو جهش یافته است، مطالعاتی که پروفایلهای جهشی را بررسی میکنند، نشان دادهاند که آنها بین CRC و SBA متفاوت هستند، که نشان میدهد مسیر آدنوکارسینوم بین آدنوکارسینومهای روده کوچک و بزرگ یکسان نیست.
جهشهای رده زایا در ژن APC در پولیپ آدنوماتوز خانوادگی (FAP) یافت میشوند. بیماران مبتلا به FAP در ۸۰٪ موارد دچار آدنومهای دوازدهه میشوند که ۴٪ آنها به آدنوکارسینوم مبتلا میشوند (واسن و همکاران). به دلیل خطر تبدیل به بدخیمی، نظارت آندوسکوپی مورد نیاز است.
سندرم لینچ با غیرفعال شدن سلولهای زایا در یک یا چند ژن ترمیم عدم تطابق (MLH1، MSH2، MSH6 و PMS2) همراه است که منجر به بیثباتی ریزماهوارهها میشود. این غیرفعال شدن در بیش از ۹۰٪ از بیماران مبتلا به سندرم لینچ یافت میشود. جهشهای پراکنده، به ویژه در MLH1، همچنین میتوانند با غیرفعال شدن کلی سیستم ترمیم عدم تطابق (MMR) که در ۱۵٪ از موارد CRC رخ میدهد، رخ دهند. SBA در تا ۳۵٪ موارد کمبود MMR (dMMR) دارد که بسیار بیشتر از آدنوکارسینوم کولورکتال است، که بار دیگر نشان میدهد مکانیسمهای مولکولی پشت توسعه SBA با آدنوکارسینومهای روده بزرگ متفاوت است (اورمن و همکاران). بیماران SBA مبتلا به dMMR بیشتر احتمال دارد که ضایعاتشان در قسمت پروگزیمال (دوازدهه، ژژنوم) به جای قسمت دیستال روده کوچک، متمرکز باشد. پروتئینهای مهم دیگری که اغلب در SBA جهش مییابند، KRAS و p53 هستند که میزان جهش آنها به ترتیب 53٪ و 52٪ است.
مرحلهبندی
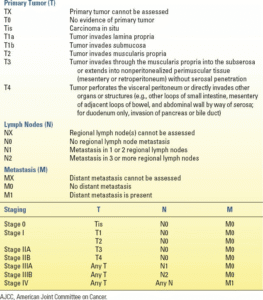
مرحلهبندی ویرایش هشتم کمیته مشترک سرطان آمریکا (AJCC) برای آدنوکارسینوم روده کوچک در جدول 10.1 نشان داده شده است.
سیر بالینی
علائم بالینی SBA اغلب غیر اختصاصی هستند، بنابراین بیش از نیمی از بیماران در مرحله III یا IV بیماری مراجعه میکنند. پیشآگهی بر اساس مرحله بیماری است و میزان بقای کلی 5 ساله برای مرحله I، 50٪ تا 60٪، برای مرحله II، 39٪ تا 55٪، برای مرحله III، 10٪ تا 40٪ و برای مرحله IV، 3٪ تا 5٪ است (Aparicio و همکاران). در مقایسه با سایر بدخیمیهای روده کوچک مانند تومورهای کارسینوئید، لنفوم و استرومال، SBA پیشآگهی ضعیفتری دارد.
متاستاز غدد لنفاوی در زمان تشخیص در 35٪ از بیماران وجود دارد و 20٪ بیماری دوردست خواهند داشت که کبد شایعترین محل آن است. یک مطالعه پایگاه داده SEER توسط Overman و همکارانش که ارزش پیشآگهی غدد لنفاوی در SBA را بررسی میکرد، نشان داد که با تعداد کل بیشتر غدد لنفاوی بررسی شده برای بیماری مرحله I-III، بقای بیشتری وجود دارد. علاوه بر این، در بیماران مبتلا به بیماری مرحله III، بقای 5 ساله اختصاصی بیماری ضعیفتری وجود داشت، زمانی که ≥3 غدد لنفاوی مثبت در مقایسه با <3 (37٪ در مقابل 58٪) یافت شد.
نشانگرهای تومور برای SBA مشابه آدنوکارسینوم روده بزرگ هستند و شامل آنتیژن کارسینوامبریونیک (CEA) و آنتیژن کربوهیدرات 19-9 (CA 19-9) میشوند. CEA و CA 19-9 به ترتیب در 30٪ و 40٪ بیماران افزایش یافته است (Raghav و همکاران). این نشانگرها میتوانند به عنوان عوامل پیشآگهی برای بقای بدون پیشرفت/کلی و برای نظارت بر عود بیماری استفاده شوند.
درمان جراحی
درمان اولیه برای SBA شامل برداشتن کامل تومور با لنفادنکتومی موضعی-منطقهای است. بیماران مبتلا به بیماری غیرقابل برداشت، بقای کلی 5 ساله 0٪ در مقایسه با 54٪ برای بیماری قابل برداشت دارند. ضایعات پروگزیمال دوازدهه با پانکراتیکودئودنکتومی همراه با لنفادنکتومی موضعی-منطقهای درمان میشوند.
برای ضایعات انتهایی دوازدهه، برداشتن وسیع همراه با لنفادنکتومی ممکن است مناسب باشد، به شرطی که حاشیههای منفی به دست آید. ضایعات ژژنوم و ایلئوم معمولاً با برداشتن وسیع و تشریح غدد لنفاوی ناحیهای درمان میشوند. برای ضایعات ایلئوم نزدیک به سکوم، ممکن است برای به دست آوردن حاشیههای منفی، کولکتومی راست یا ایلئوسککتومی لازم باشد.
| جدول10.1 مرحلهبندی سرطان روده کوچک، به استثنای لنفوم، کارسینوئید و سارکومهای احشایی، ویرایش هشتم AJCC |
 |
درمان کمکی
عود پس از برداشتن تومور بدخیم ساباورال میتواند تا 39٪ باشد که به استدلال برای درمان کمکی اعتبار میبخشد. از بین موارد عود، 56٪ دوردست، 19٪ کارسینوماتوز و 17٪ موضعی هستند (دباجا و همکاران). دادههای تصادفی آیندهنگر که رژیم درمانی کمکی بهینه برای ساباورال را بررسی کنند، کم است و توصیههای فعلی عمدتاً بر اساس دادههای گذشتهنگر هدایت میشوند. با وجود کمبود دادههای با کیفیت بالا، یک مطالعه پایگاه داده ملی سرطان نشان داد که میزان استفاده از شیمیدرمانی کمکی از 8٪ در سال 1985 به 22٪ در سال 2005 افزایش یافته است (p < 0.0001). بر اساس دادههای موجود، شیمیدرمانی مبتنی بر 5-فلورواوراسیل استاندارد برای بیماران مبتلا به ساباورال با ریسک بالا (بهعلاوه غدد لنفاوی) است (راگاو و همکاران). برای رسیدگی به این کمبود دادهها برای درمان کمکی، ابتکار بینالمللی سرطانهای نادر (IRCI) ایجاد شد. این یک ابتکار مشترک بین تحقیقات سرطان بریتانیا (CRUK)، شبکه تحقیقات بالینی موسسه ملی سلامت: سرطان (NIHR CRN:Cancer)، موسسه ملی سرطان (NCI)، سازمان اروپایی تحقیقات و درمان سرطان (EORTC)، موسسه ملی سرطان (INCa) و گروه کارآزماییهای بالینی موسسه ملی سرطان کانادا (NCIC CTG) است که هدف آن «تسهیل توسعه کارآزماییهای بالینی بینالمللی برای بیماران مبتلا به سرطانهای نادر به منظور افزایش پیشرفت درمانهای جدید برای این بیماران» است. برای SBA، مطالعه BALLARD یک کارآزمایی تصادفی آیندهنگر بزرگ است که به طور خاص تأثیر شیمیدرمانی کمکی را برای آدنوکارسینومهای روده کوچک ارزیابی میکند (Keat و همکاران).
HIPEC
شیمی درمانی داخل صفاقی هیپرترمیک (HIPEC) همراه با جراحی سیتورداکتیو (CRS) استاندارد مراقبت برای سودومیکسوما پریتونئی، مزوتلیومای پریتونئال و سرطان کولورکتال با متاستازهای فقط پریتونئال است، اما به عنوان درمانی برای بیماران بسیار منتخب مبتلا به SBA در حال ظهور است. در مطالعهای که عوامل پیشآگهی SBA از MDACC را بررسی میکرد، مشخص شد که 25٪ از بیماران مبتلا به بیماری مرحله IV، کارسینوماتوز صفاقی دارند. برای این گروه بسیار انتخابی از بیماران، HIPEC/CRS نقش بالقوهای در درمان دارد. این تکنیک شامل برداشتن تمام ضایعات صفاقی قابل مشاهده و تزریق شیمی درمانی گرم به حفره شکم است.
در یک مطالعه چند موسسهای از هلند توسط ون اودهوسدن و همکارانش که HIPEC/CRS را برای SBA بررسی میکردند، برداشتن کامل در 93.8٪ با میانگین بقای کلی 30.8 ماه در مقابل 7.1 ماه در بیمارانی که تحت عمل جراحی قرار نگرفتند، به دست آمد. در MDACC، الگوریتم درمان برای بیماران مبتلا به SBA متاستاتیک با بیماری فقط صفاقی به شرح زیر است: (1) بیماران در یک هیئت تومور چند رشتهای با ارزیابی وسعت بیماری مورد بحث قرار میگیرند، (2) شیمیدرمانی سیستمیک آغاز میشود، (3) ارزیابی پیشرفت از طریق تصویربرداری مقطعی و/یا لاپاراسکوپی تشخیصی، و (4) اگر پیشرفتی وجود نداشته باشد و بیماری محدود قابل برداشتن کامل باشد، HIPEC با CRS ارائه میشود.
بیماری متاستاتیک
شیمیدرمانی سیستمیک درمان اصلی برای بیماران مبتلا به SBA متاستاتیک است. دادههای حاصل از مطالعات گذشتهنگر، بهبود بقای کلی با شیمیدرمانی سیستمیک را 13 ماه در مقابل 4 ماه نشان دادهاند (p = 0.02) (Czaykowski و همکاران). این مطالعات از طیف گستردهای از داروها استفاده کردهاند، اما درمان استاندارد باید شامل استفاده از CAPOX (کاپسیتابین و اگزالیپلاتین) یا FOLFOX (5-فلوروراسیل، لوکوورین و اگزالیپلاتین) باشد. در حال حاضر، کارآزماییهای بالینی در حال بررسی استفاده از این ستون فقرات با بواسیزوماب، ایرینوتکان، پانیتوموماب، ارلوتینیب و پاکلیتاکسل برای کمک به تعیین درمان بهینه برای SBA متاستاتیک هستند.
تومورهای کارسینوئید
اپیدمیولوژی
تومورهای نورواندوکرین (NETs) گروه متنوعی از تومورها هستند که از سلولهای انتروکرومافین موجود در سراسر بدن ناشی میشوند. آنها میتوانند از روده قدامی، روده میانی یا روده خلفی ناشی شوند و توانایی ترشح هورمونها و پپتیدهای مختلف را دارند که منجر به سندرمهای عملکردی میشود.
حداکثر بروز تومورهای کارسینوئید در دهههای هفتم تا هشتم زندگی است. در مورد NETهای ژژنوم/ایلئال، میزان بالاتری در مردان در مقایسه با زنان رخ میدهد (0.8 در مقابل 0.57 در هر 100000) (یائو و همکاران).
کارسینوئیدهای روده کوچک
42٪ از NETهای دستگاه گوارش را تشکیل میدهند و بیشتر از ایلئوم انتهایی ناشی میشوند. به طور کلی، میزان بروز کارسینوئیدهای روده کوچک در حال افزایش است و بر اساس دادههای SEER از سال ۱۹۷۳ تا ۲۰۰۴، از ۲.۱ به ۹.۳ در هر میلیون نفر افزایش یافته است. تومورهای NET روده کوچک از SBA به عنوان شایعترین تومور اولیه روده کوچک پیشی گرفتهاند.
عوامل خطر برای ایجاد کارسینوئیدهای SB شامل سابقه خانوادگی سرطان پروستات یا روده بزرگ در بین بستگان درجه یک؛ سرطان زبان یا دهان در خواهر و برادر؛ و لنفوم آندومتر، کلیه، پوست یا لنفوم غیر هوچکین در والدین است (Boudreaux و همکاران).
عوامل خطر محیطی برای ایجاد تومور کارسینوئید نامشخص است. در یک مطالعه مورد-شاهدی مبتنی بر جمعیت اروپایی، سیگار کشیدن خطر ابتلا به کارسینوئید روده کوچک را افزایش داد، اما در یک مطالعه مورد-شاهدی مبتنی بر ایالات متحده اینطور نبود، در حالی که مصرف الکل در هیچ یک از مطالعات این خطر را افزایش نداد.
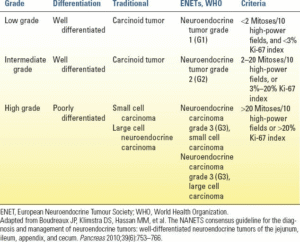
| جدول10.2 سیستم درجهبندی تومور نورواندوکرین (NET) روده میانی |
 |
آسیبشناسی
سیستمهای نامگذاری متعددی هنگام توصیف تومورهای کارسینوئیدی (NETs) وجود دارد که به سردرگمی میافزاید و کاربرد دادهها را در هنگام استفاده از سیستمهای مختلف محدود میکند.
در تلاش برای استانداردسازی اصطلاحات، انجمن تومور نورواندوکرین اروپا (ENETS) و سازمان بهداشت جهانی معیارهایی را تعیین کردند تا اطمینان حاصل شود که پزشکان به یک زیرگروه تومور اشاره میکنند. بر اساس این معیارها، تومورهای کارسینوئیدی روده کوچک بر اساس تعداد میتوز و شاخص تکثیر Ki-67 به سه درجه تقسیم میشوند (جدول 10.2).
اصطلاح “تومور کارسینوئید” به طور خاص به تومورهای درجه پایین/متوسط، تومورهای تمایز یافته یا تومورهای نورواندوکرین درجه 1 و 2 (همچنین تومورهای نادر تمایز یافته درجه 3) اشاره دارد.
به طور کلی، در زمان مراجعه، درگیری غدد لنفاوی منطقهای در 70٪ از بیماران و متاستاز کبدی در بیش از نیمی از آنها مشاهده میشود. ندولهای زیر مخاطی، و قادر به آزاد کردن مواد فعال بیولوژیکی مانند سروتونین، آمینها و پروستاگلاندینها هستند. از آنجا که اغلب چندین تومور یافت میشوند، ارزیابی دقیق کل روده کوچک در طول عمل ضروری است.
مرحلهبندی
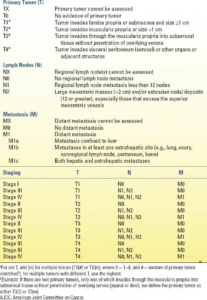
مرحلهبندی نسخه هشتم کمیته مشترک سرطان آمریکا (AJCC) برای تومورهای کارسینوئید در جدول 10.3 نشان داده شده است.
| جدول10.3 مرحلهبندی ویرایش هشتم AJCC برای روده کوچک تومورهای «کارسینوئید» (تومورهای نورواندوکرین) درجه I و II، و درجه III نادر با تمایز خوب منشعب از ژژونوم و ایلئوم |
 |
تظاهرات بالینی
کارسینوئیدهای روده کوچک معمولاً کند هستند؛ این میتواند منجر به تأخیر در تشخیص شود و بسیاری از بیماران به طور متوسط 5 سال قبل از تشخیص، علائم را دارند. مانند SBA، اکثر این بیماران با درد مزمن شکمی و/یا انسداد غیر اختصاصی مراجعه میکنند.
بیست تا سی درصد از بیماران میتوانند با سندرم کارسینوئید مراجعه کنند. علائم سندرم کارسینوئید شامل گرگرفتگی (94٪)، اسهال آبکی (80٪)، خس خس سینه، درد شکم و پلاگر (کمبود نیاسین) است. گرگرفتگی میتواند با مصرف الکل، پنیر آبی، شکلات یا شراب قرمز ایجاد شود. سندرم کارسینوئید طولانی مدت در نهایت میتواند منجر به ایجاد نارسایی قلب سمت راست (بیماری قلبی کارسینوئید) شود.
شدیدترین تظاهر سندرم کارسینوئید، بحران کارسینوئید است. علائم شامل بیثباتی همودینامیک، شوک غیرپاسخگو به داروهای منقبض کننده عروق، هایپرترمی، آریتمی و/یا انقباض برونش است. افرادی که بیشترین خطر ابتلا به بحران کارسینوئید را دارند، بیمارانی هستند که دچار گرگرفتگی و تومورهای بزرگ و حجیم هستند. بحران کارسینوئید میتواند با جراحی ایجاد شود و بنابراین به همه بیماران مبتلا به NET باید قبل از عمل اکتروتاید داده شود.
تشخیص
تشخیص تومورهای کارسینوئید با تصویر بالینی، آزمایش بیوشیمیایی و تصویربرداری انجام میشود.
کروموگرانین A (CgA) یک پلیپپتید موجود در گرانولهای ترشحی سلولهای نورواندوکرین است. CgA در 60 تا 100 درصد از بیماران مبتلا به NET های عملکردی یا غیرفعال افزایش مییابد و به ترتیب حساسیت و اختصاصیت 70 و 100 درصد دارد. افزایش کاذب CgA را میتوان در نارسایی کلیوی/کبدی، بیماران مبتلا به گاستریت مزمن و افرادی که مهارکنندههای پمپ پروتون (PPI) مصرف میکنند، مشاهده کرد. مصرف PPI ها باید 2 هفته قبل از ارزیابی CgA متوقف شود. یکی از هورمونهای اصلی آزاد شده توسط تومورهای کارسینوئید، سروتونین است که توسط کبد به 5-هیدروکسی ایندول استیک اسید (5-HIAA) متابولیزه شده و سپس به ادرار دفع میشود. اندازهگیری مستقیم سطح سروتونین سرم میتواند دقیق نباشد زیرا تغییرات روزانه قابل توجهی بر اساس استرس و سطح فعالیت وجود دارد. سطح 5-HIAA ادرار بیست و چهار ساعته، اندازهگیری دقیقتری از فعالیت سروتونین سرم ارائه میدهد.
افزایش سطح در بیش از نیمی از بیماران مشاهده میشود و اختصاصیت آن برای تومورهای کارسینوئید تا 88٪ است. با این حال، مانند CgA، 5-HIAA میتواند به طور کاذب افزایش یابد و نتایج نادرستی پس از مصرف اخیر الکل، آجیل، گوجه فرنگی و داروهای سرماخوردگی/سرفه مشاهده میشود (تورمی و همکاران). از آنجا که سطح CgA و 5-HIAA میتوانند منعکس کننده بار کلی تومور باشند، میتوان آنها را برای تشخیص عود تومور پس از برداشتن اولیه تومور دنبال کرد. در شرایطی که 5-HIAA بالا نباشد، آزمایشهای اضافی باید شامل اندازهگیری 5-هیدروکسیتریپتامین (5-HT، سروتونین) و 5-هیدروکسیتریپتوفان (5-HPT) در ادرار، 5-HPT پلاسما، 5-HT پلاکتی و سطح سرمی CgA، انولاز اختصاصی نورون (NSE)، ماده P و نوروپپتید K باشد.
تصویربرداری مقطعی با سیتیاسکن یا امآرآی میتواند تومورهای کارسینوئید روده کوچک را با حساسیت 77 تا 80 درصد شناسایی کند. تومورهای کارسینوئید روده کوچک ضایعات عروقی هستند و تصویربرداری سیتیاسکن باید با برشهای نازک با فاز شریانی و وریدی انتهایی انجام شود تا بتوان افزایش شدت شریانی و شستشوی وریدی را شناسایی کرد. این امر به ویژه در موارد متاستاز کبدی مفید است. در امآرآی، تومورهای کارسینوئید میتوانند در تصاویر T1 ظاهری متغیر داشته باشند، به صورت هیپواینتنس یا ایزواینتنس، در حالی که متاستازهای کبدی معمولاً در تصاویر T2 روشن به نظر میرسند. با هر روش تصویربرداری مورد استفاده، تقریباً 25٪ از بیماران وسعت بیماری را کمتر از حد واقعی تخمین میزنند (چمبرز و همکاران).
اکترئواسکن میتواند به عنوان کمکی برای سیتیاسکن یا امآرآی در شناسایی بیماریهای موضعی-منطقهای یا دوردست استفاده شود. این روش حساسیتی تا 90٪ دارد و برخلاف تصویربرداری مقطعی موضعی، کل بدن را برای ضایعات پنهان بررسی میکند. اکترواسکن گیرندههای سوماتوستاتین 2 و 5 را در تومورها شناسایی میکند. در برخی موارد، بیمارانی که NET های تمایز نیافته دارند، این گیرندهها را از دست دادهاند و این ضایعات با اکترواسکن قابل تشخیص نیستند.
درمان بیماریهای موضعی
برداشتن کامل تومور اولیه با لنفادنکتومی منطقهای، درمان استاندارد برای بیماریهای موضعی است. قبل از هر عمل جراحی، بیماران دارای علائم سندرم کارسینوئید باید با اکتروتاید زیر جلدی (250 تا 500 میکروگرم) درمان شوند تا از بروز بحران کارسینوئید جلوگیری شود (پاریس و همکاران). دوزهای اضافی باید در زمان دستکاری تومور در دسترس باشند تا تجویز شوند.
درگیری غدد لنفاوی ناحیهای در ۷۰٪ از بیماران مشاهده میشود؛ بنابراین، برداشتن مزانتر نه تنها برای اهداف برداشتن کامل، بلکه برای کنترل موضعی نیز الزامی است. این امر میتواند چالش برانگیز باشد زیرا کارسینوئیدهای روده کوچک میتوانند واکنش دسموپلاستیک قابل توجهی ایجاد کنند که منجر به انقباض و کوتاه شدن مزانتر میشود. هنگامی که برداشتن مزانتر امکانپذیر است اما انجام نمیشود، غدد لنفاوی باقی مانده در نهایت میتوانند به دلیل ترومبوز عروق مزانتر باعث انسداد یا ایسکمی روده شوند. از آنجایی که کارسینوئیدهای روده کوچک اغلب چند مرکزی هستند، باید کل روده کوچک از نظر بیماری بررسی شود.
با توجه به خطر کلستاز و سنگ کیسه صفرا مرتبط با این دارو، باید انجام کوله سیستکتومی پیشگیرانه در بیمارانی که به مدت طولانی آنالوگهای سوماتوستاتین مصرف میکنند، در نظر گرفته شود.
در بیمارانی که تحت عمل جراحی برداشتن کامل تومورهای کارسینوئید روده کوچک موضعی و بدون گره لنفاوی قرار میگیرند، میانگین بقای کلی 111 ماه است، در حالی که در افراد مبتلا به بیماری منطقهای 105 ماه است (یائو و همکاران).
درمان بیماری پیشرفته
برداشتن تومورهای کارسینوئید حتی در شرایط بیماری متاستاتیک ممکن است در تسکین، تسکین علائم و کنترل موضعی-منطقهای نقش داشته باشد.
اگر بیماری متاستاتیک وجود داشته باشد و برداشتن کامل همه ضایعات امکانپذیر باشد، باید تلاش شود تا همه بیماریها به طور کامل ریشهکن شوند، زیرا میتواند بقای بدون بیماری را طولانیتر کرده و تسکین علائم را فراهم کند. در بیماران مبتلا به متاستازهای کبدی، در 82٪ از بیماران پس از برداشتن همه ضایعات، تسکین نسبی یا کامل علائم حاصل میشود. همانند بیماری موضعی، قبل از عمل باید اکترئوتاید تجویز شود تا از بحران کارسینوئید جلوگیری شود و در صورت نیاز به اکترئوتاید طولانی مدت، کوله سیستکتومی پیشگیرانه در نظر گرفته شود.
بیماران مبتلا به بیماری متاستاتیک غیرقابل جراحی که بدون علامت هستند، باید تحت نظر باشند. در صورت بروز عوارض مرتبط با تومور مانند خونریزی، انسداد یا سوراخ شدن، میتوان رزکسیون تسکینی را پیشنهاد کرد.
برای بیماران علامتدار مبتلا به متاستازهای کبدی که نمیتوان آنها را به طور کامل برداشت و به مدیریت پزشکی مقاوم هستند، درمانهای تخریبی هدایتشده توسط کبد یا کاهش حجم جراحی میتواند به تسکین علائم کمک کند.
آنالوگهای سوماتوستاتین را میتوان برای تسکین علائم و تثبیت بیماری استفاده کرد. مطالعه PROMID، یک کارآزمایی تصادفی کنترلشده دوسوکور، اکترئوتاید LAR (رهش طولانیاثر) را در مقایسه با دارونما در بیماران بدون درمان با NETهای روده میانی مقایسه کرد. این مطالعه نشان داد که درمان با اکتروتاید LAR میانگین زمان پیشرفت را طولانیتر میکند (14.3 ماه در مقابل 6.0 ماه) (رینکه و همکاران). در بررسی دیگری از 15 مطالعه توسط مادلین و همکاران که درمان NETها را با اکتروتاید LAR بررسی میکردند، بهبود علائم در 74٪ و پاسخ تومور در 70٪ مشاهده شد.
برای بیمارانی که علائم مقاوم به آنالوگهای سوماتوستاتین دارند، نشان داده شده است که اینترفرون آلفا (IFNα) در 50٪ موارد اسهال را کاهش میدهد، با این حال، این تسکین گذرا بود.
درمانهای ترکیبی با آنالوگهای سوماتوستاتین و شیمیدرمانی سیستمیک مورد بررسی قرار گرفتهاند. کارآزمایی RADIANT-2 یک مطالعه فاز 3، دوسوکور و کنترلشده با دارونما در بیماران مبتلا به NETهای پیشرفته و سندرم کارسینوئید بود که اثر اورولیموس، یک مهارکننده هدف پستانداران راپامایسین (mTOR) را ارزیابی میکرد. بیماران به صورت تصادفی به دو گروه تقسیم شدند: گروه اول، اورولیموس به همراه اکتروتاید LAR یا گروه دوم، دارونما و اکتروتاید LAR. این مطالعه بهبود در بقای بدون پیشرفت بیماری را از 11.3 ماه به 16.4 ماه برای گروه دوم که اورولیموس به همراه اکتروتاید LAR دریافت میکردند، نشان داد (پاول و همکاران).
در مقایسه با آدنوکارسینوم پیشرفته روده کوچک، تومورهای کارسینوئید پیشآگهی بهتری دارند و میزان بقای کلی 5 ساله آنها 68٪ با بیماری متاستاتیک برداشته شده و 38٪ با بیماری متاستاتیک غیرقابل برداشت است.
سارکوم
اپیدمیولوژی/پاتولوژی
سارکومهای روده کوچک 8٪ از کل بدخیمیهای اولیه روده کوچک و 2٪ از کل سارکومها را تشکیل میدهند (هاو و همکاران). آنها اغلب تومورهایی با رشد آهسته هستند که شایعترین انواع آنها لیومیوسارکوم و تومور استرومایی دستگاه گوارش (GIST) هستند که 75٪ از سارکومهای روده کوچک را تشکیل میدهند. فیبروسارکوم، لیپوسارکوم و آنژیوسارکوم زیرگروههای کمتر شایعی هستند. بر اساس یک مطالعه NCDB، ۴۶.۸٪ از سارکومهای روده کوچک در ژژونوم، ۲۷.۷٪ در دوازدهه و ۲۵.۵٪ در ایلئوم قرار دارند. این تومورها میتوانند از طریق خون به کبد، ریهها و استخوان گسترش یابند و قادر به تهاجم به بافتهای مجاور هستند.
عوامل خطر مرتبط با ایجاد سارکومهای روده کوچک عبارتند از:
ویروس اپشتین بار (EBV)، پرتودرمانی، رتینوبلاستومای ارثی و سندرم لی-فرومنی.
تظاهرات بالینی
بیماران معمولاً با درد شکمی غیر اختصاصی (۶۵٪)، توده قابل لمس (۵۰٪) و خونریزی دستگاه گوارش مراجعه میکنند. از آنجا که سارکومها به طور کلی رشد آهستهای دارند، بیش از ۷۵٪ از ضایعات در زمان تشخیص بیش از ۵ سانتیمتر اندازه دارند و بیش از نیمی از بیماران با گسترش منطقهای یا متاستازهای دور مراجعه میکنند. تصویربرداری مقطعی اغلب تودهای ناهمگن را نشان میدهد که نکروز در آن وجود دارد، اگر تومور از منبع خونرسانی فراتر رفته باشد.
درمان
جراحی با برداشتن کامل تومور، همچنان درمان اصلی سارکومهای روده کوچک است. برخلاف آدنوکارسینوم روده کوچک و تومورهای کارسینوئید، سارکومها به ندرت به غدد لنفاوی مزانتریک متاستاز میدهند و بنابراین لنفادنکتومی گسترده لازم نیست. در بیمارانی که بیماری متاستاتیک و عوارض خونریزی یا انسداد دارند، برداشتن تسکینی یا بای پس روده باید برای کنترل علائم در نظر گرفته شود.
شیمیدرمانی سیستمیک و شیمیدرمانی-پرتودرمانی نقش محدودی در درمان سارکومهای روده کوچک دارند. در مورد لیومیوسارکومها، 50٪ دچار عود موضعی و/یا متاستاز دوردست میشوند (Shenoy و همکاران).
گزینههای درمانی غیرجراحی با شیمیدرمانی یا شیمیدرمانی-پرتودرمانی ضعیف هستند، زیرا لیومیوسارکومها در برابر پرتودرمانی نسبتاً مقاوم هستند و شیمیدرمانیهای سیتوتوکسیک سیستمیک، میزان پاسخ متغیری را با بهبود ضعیف در بقای کلی نشان دادهاند.
درمان تومورهای دستگاه گوارش موضعی و متاستاتیک به طور جداگانه مورد بحث قرار خواهد گرفت (فصل 5).
لنفوم
اپیدمیولوژی/پاتولوژی
روده کوچک دومین محل شایع لنفوم اولیه دستگاه گوارش پس از معده است و 15٪ از کل بدخیمیهای اولیه روده کوچک را تشکیل میدهد. لنفومهای دستگاه گوارش میتوانند از سلولهای B یا T ناشی شوند و شامل لنفومهای بورکیت، لنفومهای بافت لنفاوی مرتبط با مخاط (MALT)، لنفومهای سلول B بزرگ منتشر و لنفومهای سلول مانتل میشوند. عوامل خطر مرتبط با ایجاد لنفوم روده کوچک شامل بیماری سلیاک، ویروس نقص ایمنی انسانی (HIV)، اختلال لنفوپرولیفراتیو پس از پیوند (PTLD)، هلیکوباکتر پیلوری و بیماری ایمونوپرولیفراتیو روده کوچک (IPSID) است.
لنفومهای روده کوچک از بافتهای لنفاوی ناشی میشوند و بیشترین تراکم آنها در ایلئوم (60٪ تا 65٪) و پس از آن در ژژونوم (20٪ تا 25٪) و دوازدهه (6٪ تا 8٪) است. آنها اغلب چند کانونی هستند و ضایعات پراکنده در حداکثر 15٪ موارد وجود دارد (ناکامورا و همکاران).
لنفومهای روده کوچک میتوانند از طریق مخاط نفوذ کرده و منجر به زخم و خونریزی شوند، در حالی که بزرگ شدن آنها میتواند باعث انسداد شود. تقریباً 25٪ از بیماران با سوراخ شدن مراجعه میکنند.
درمان
بیمارانی که علائم لنفوم روده کوچک را ندارند، باید مشابه بیماران مبتلا به لنفوم سیستمیک درمان شوند و تحت درمان با شیمیدرمانی مانند سیکلوفسفامید، دوکسوروبیسین، وینکریستین و پردنیزون (CHOP) قرار گیرند.
بیمارانی که با عوارضی مانند خونریزی، انسداد و سوراخ شدن مراجعه میکنند، اغلب مانع از استفاده از درمان سیستمیک میشوند و در صورت عدم وجود بیماریهای همراه، نیاز به عمل جراحی دارند. در طول جراحی، ناحیه تومور باید برداشته شود و بررسی دقیق روده کوچک باقی مانده برای شناسایی ضایعات احتمالی احتمالی انجام شود. برخلاف سایر تومورهای روده کوچک، برداشتن مزانتر اختصاصی ضروری نیست، مگر اینکه برای برداشتن تومور اولیه لازم باشد. غدد لنفاوی باقی مانده پس از بهبودی بیمار با درمان سیستمیک درمان میشوند.
شیمی درمانی میتواند در بیمارانی که کاندید شیمیدرمانی سیتوتوکسیک نیستند، استفاده شود. میزان عوارض این روش به دلیل نکروز تومور، خونریزی و سوراخ شدن احتمالی روده، بالا است.
بدخیمیهای متاستاتیک
ضایعات متاستاتیک به روده کوچک شایعترین بدخیمی روده کوچک هستند. این متاستازها میتوانند از طریق مجاری خونی یا لنفاوی به روده کوچک گسترش یابند. تومورهای اولیه که اغلب به روده کوچک گسترش مییابند، از روده بزرگ، تخمدان، ریه و ملانوما ناشی میشوند.
متاستازها به طور مشخص در سطح روده کوچک یافت میشوند و میتوانند باعث انسداد یا سوراخ شدن روده شوند. ملانوما از این نظر منحصر به فرد است که میتواند به غدد لنفاوی مزانتریک گسترش یابد و بنابراین در زمان برداشتن، لنفادنکتومی موضعی توصیه میشود.
برای متاستازهای سایر تومورهای اولیه، برداشتن ضایعه بدون نیاز به انجام تشریح غدد لنفاوی منطقهای کافی است.
تسکین
ضایعات متاستاتیک به روده کوچک اغلب حجیم هستند و با برداشتن قابل اصلاح نیستند. با این حال، جراحی تسکینی در این بیماران نقش دارد. کسانی که با انسداد روده کوچک مراجعه میکنند، میتوانند کاندیداهای بالقوه بایپس رودهای باشند.
در موارد خونریزی دستگاه گوارش، آنژیوگرافی با آمبولیزاسیون انتخابی میتواند در نظر گرفته شود. با این حال، یکی از عوارض اصلی آمبولیزاسیون، ایسکمی و سوراخ شدن روده با تأخیر است و بیمارانی که تحت این عمل قرار گرفتهاند باید از نزدیک تحت نظر باشند.
بدون دیدگاه